When the Body Turns Against Itself
A Scientific Journey Through Autoimmune Disease
Scroll to explore
A Scientific Journey Through Autoimmune Disease
In autoimmune disease, the sophisticated defense system designed to protect us becomes our greatest threat. Immune cells that should respond to genuine danger signals instead react to false alarms from stressed or damaged tissues, launching a relentless attack from within.
Immune tolerance is the foundation of appropriate danger response. When tissue stress signals overwhelm regulatory capacity, autoimmunity emerges.
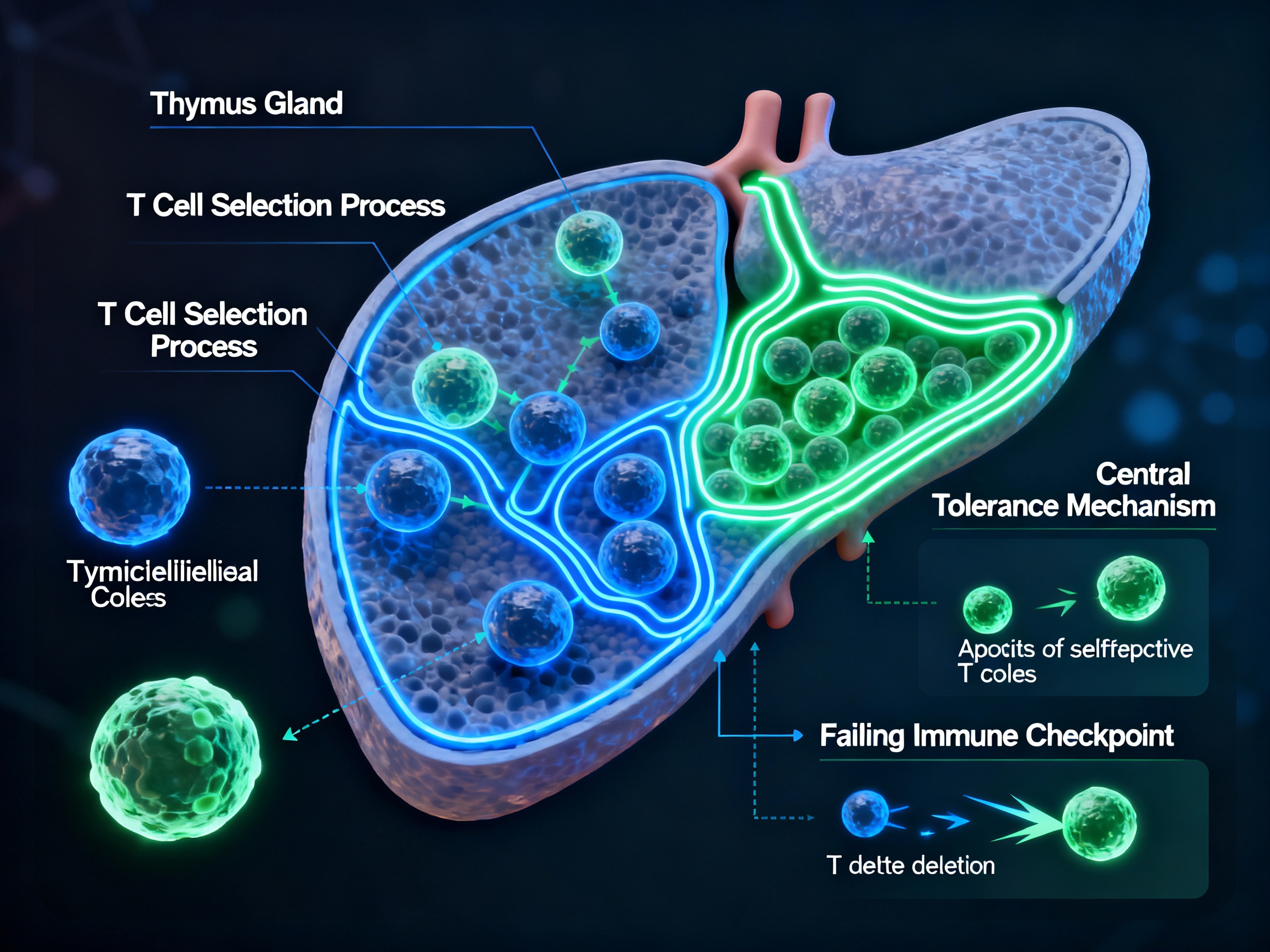
Thymic selection failure allowing autoreactive T cells to escape into circulation
Click to reveal mechanism
Negative selection of autoreactive T cells in the thymus - the primary mechanism of central immune tolerance where self-reactive T cells are eliminated during development. This process occurs in the thymic medulla where developing T cells encounter self-antigens presented by thymic epithelial cells.
Click to reveal mechanism
Clonal deletion through activation-induced cell death (AICD) and restimulation-induced cell death (RICD), immune anergy via costimulatory molecules like CTLA-4, and induction of regulatory T cells. These mechanisms serve as backup systems to eliminate or suppress autoreactive cells that escape central tolerance.
Multiple immune cell types collaborate in the autoimmune assault, each playing a distinct and devastating role.
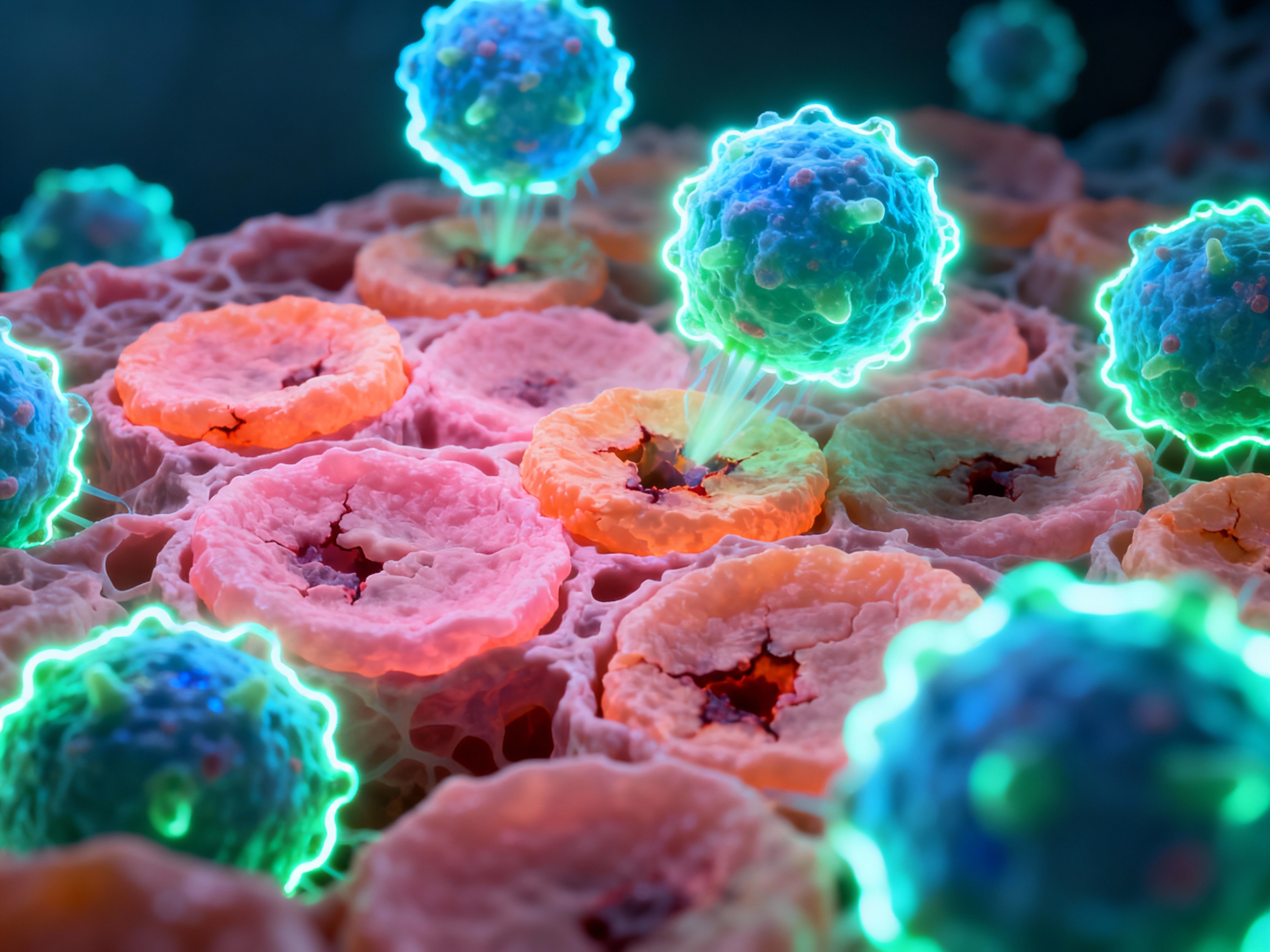
The orchestrators of autoimmune destruction
CD4+ Helper T Cells: Coordinate immune response, differentiate into Th1, Th2, Th17 subtypes, secrete inflammatory cytokines, provide B cell help. These cells amplify the autoimmune response through cytokine production.
CD8+ Cytotoxic T Cells: Directly contact and kill target cells via FasL-dependent mechanisms, promote lesion formation. They deliver lethal hits to cells displaying self-antigens.
Regulatory T Cells (Tregs): FOXP3+ cells that suppress autoreactive responses and maintain peripheral tolerance - their dysfunction leads to autoimmunity.
Antibody factories and antigen presenters
Multiple Roles: B lymphocytes produce autoantibodies targeting self-antigens, present antigens to T cells via MHC-II molecules, and secrete inflammatory cytokines including TNF-α and IL-6.
APC Function: B cells serve as professional antigen-presenting cells, capturing antigens through their surface immunoglobulins and presenting processed peptides to helper T cells, driving autoimmune responses through this bidirectional communication.
Initiators of the autoimmune cascade
Dendritic cells present self-antigens to T cells via MHC complexes, provide costimulatory signals through CD28, ICOS, and CD40 interactions. They are the bridge between innate and adaptive immunity, determining whether an immune response will be tolerogenic or inflammatory. In autoimmunity, aberrant antigen presentation drives pathogenic T cell activation.
Antigen presentation by dendritic cells to T cells via MHC complexes forms the foundation of autoimmune activation. Costimulatory pathways amplify signals through CD28-B7, ICOS, CD40-CD40L, and OX40 interactions, driving sustained autoreactive responses.
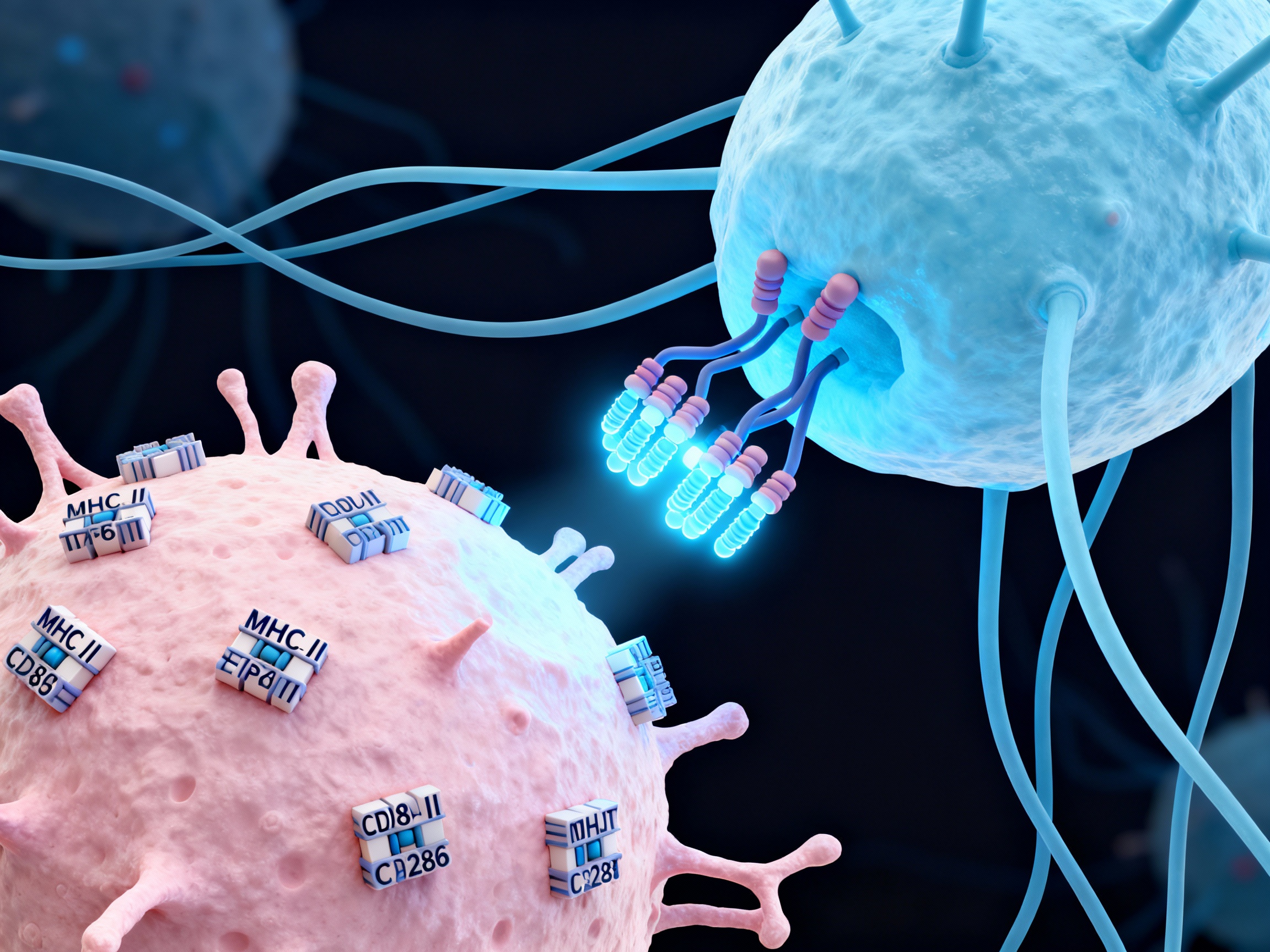
Antigen presentation by dendritic cells initiating autoreactive T cell activation via MHC-peptide complexes
Inflammatory cytokines create a cascade of tissue destruction, amplifying damage throughout the body.

Inflammatory cytokine cascade driving progressive tissue damage
Drives joint inflammation, cartilage destruction, bone erosion in RA, sensitizes pain receptors, promotes tissue degradation
Key role in inflammation, promotes B cell differentiation, acute phase response, chronic inflammation
Initiates inflammatory cascade, activates endothelial cells, recruits immune cells, causes tissue damage and pain
Produced by Th17 cells, recruits neutrophils, amplifies inflammation, drives autoimmune pathology
Type I interferon causes metabolic dysregulation in SLE, links activation to metabolic shifts, worsens disease
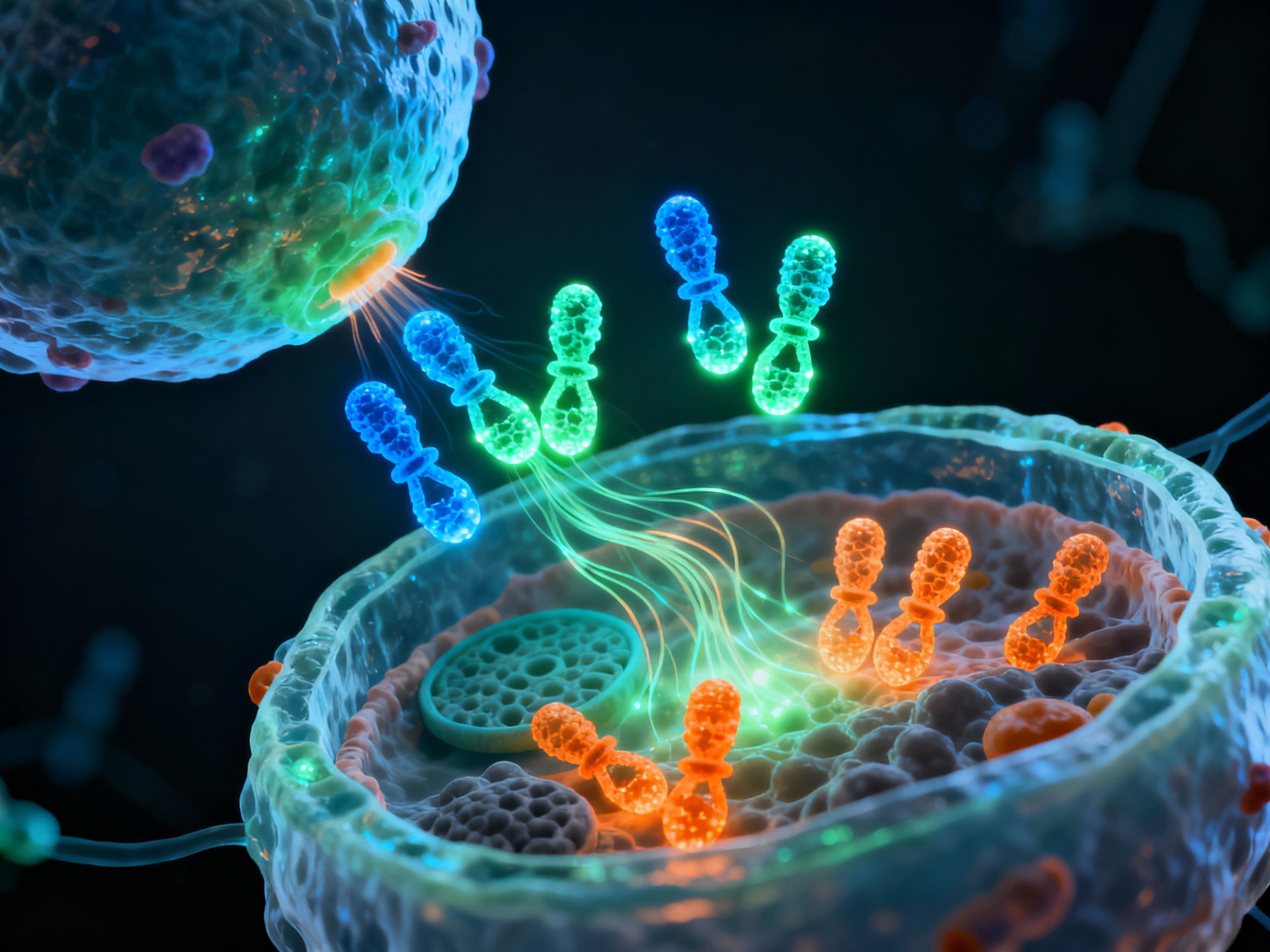
In lymphoid germinal centers, activated B cells undergo somatic hypermutation and class switching with help from follicular helper T cells, differentiating into long-lived plasma cells that continuously secrete high-affinity autoantibodies.
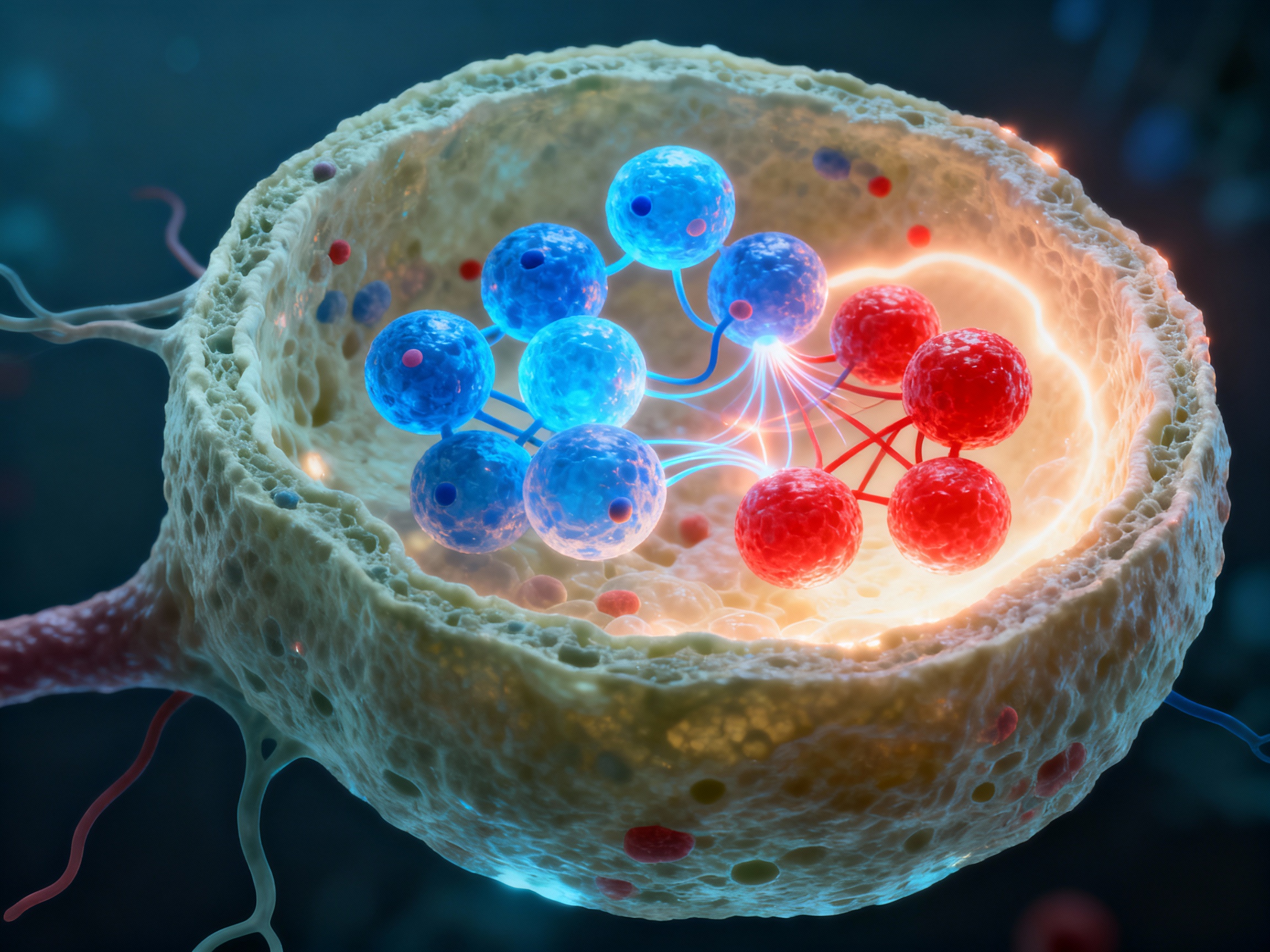
Germinal center formation - the factory for high-affinity autoantibody production
Activated B cells migrate to lymphoid follicles and form germinal centers where they undergo rapid proliferation and somatic hypermutation, creating diverse antibody variants.
Follicular helper T cells (Tfh) provide CD40L and secrete IL-21 and IL-4, critical signals that drive B cell class switching from IgM to IgG, IgA, or IgE isotypes.
Through rounds of mutation and selection, B cells with highest affinity for self-antigens are selected, creating increasingly dangerous autoantibodies that bind target tissues with lethal precision.
Selected high-affinity B cells exit the germinal center and differentiate into long-lived plasma cells that migrate to bone marrow niches. These antibody factories continuously secrete autoantibodies for months or years, forming immune complexes that activate complement cascades and directly target tissue antigens, sustaining chronic autoimmune disease even without ongoing T cell help.
Different autoimmune diseases target specific organs, each with unique mechanisms of destruction.
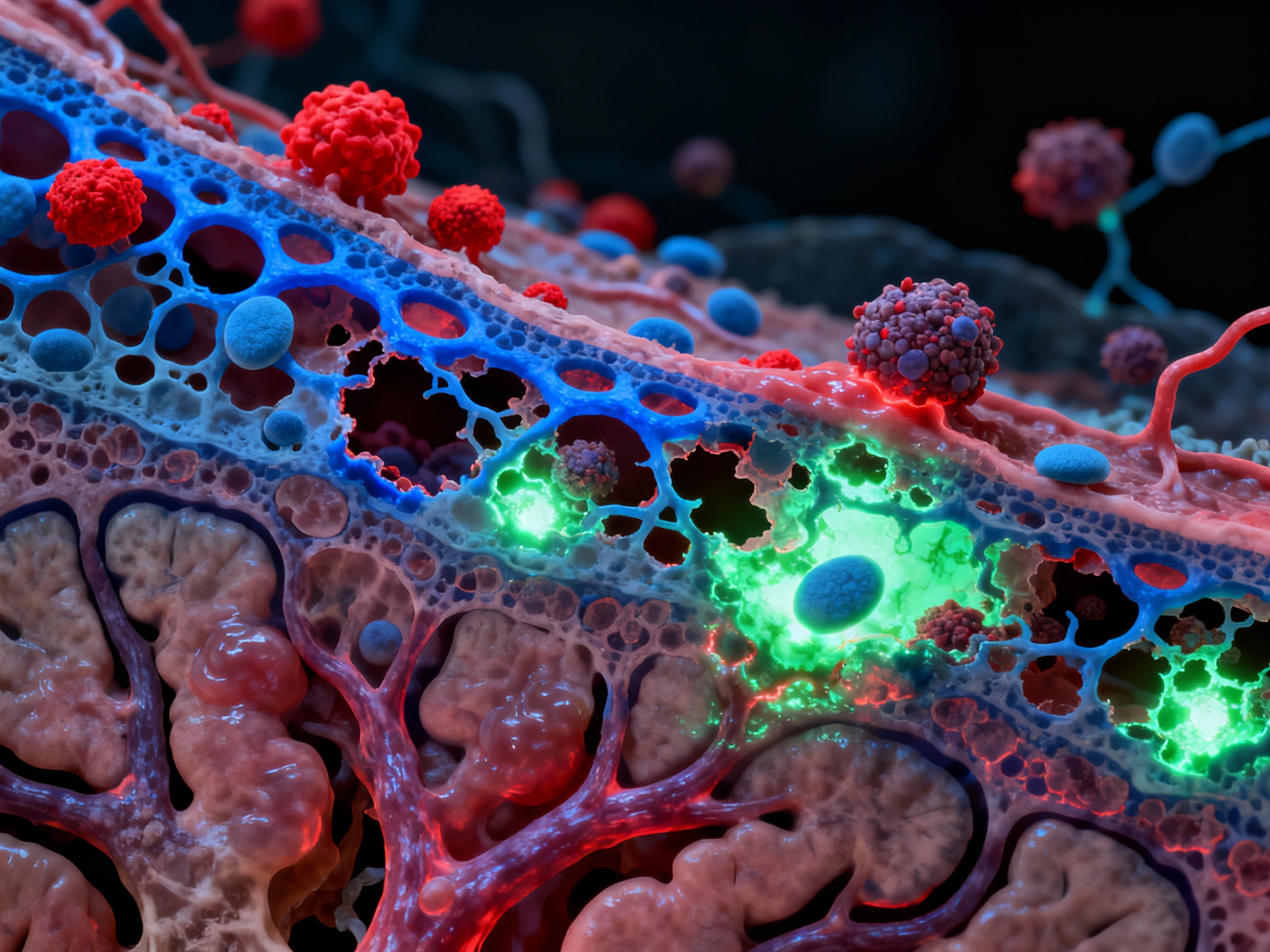
Autoreactive immune cells breaching the blood-brain barrier to attack myelin
Autoreactive T cells breach the blood-brain barrier via VCAM-1 and ICAM-1 adhesion molecules, infiltrate the central nervous system, and attack the myelin sheath of oligodendrocytes. This demyelination disrupts nerve signal transmission, causing neurological symptoms. B cells migrate into the CNS and secrete anti-myelin antibodies, amplifying the attack. The inflammatory cascade damages both myelin and the underlying axons, leading to progressive disability.
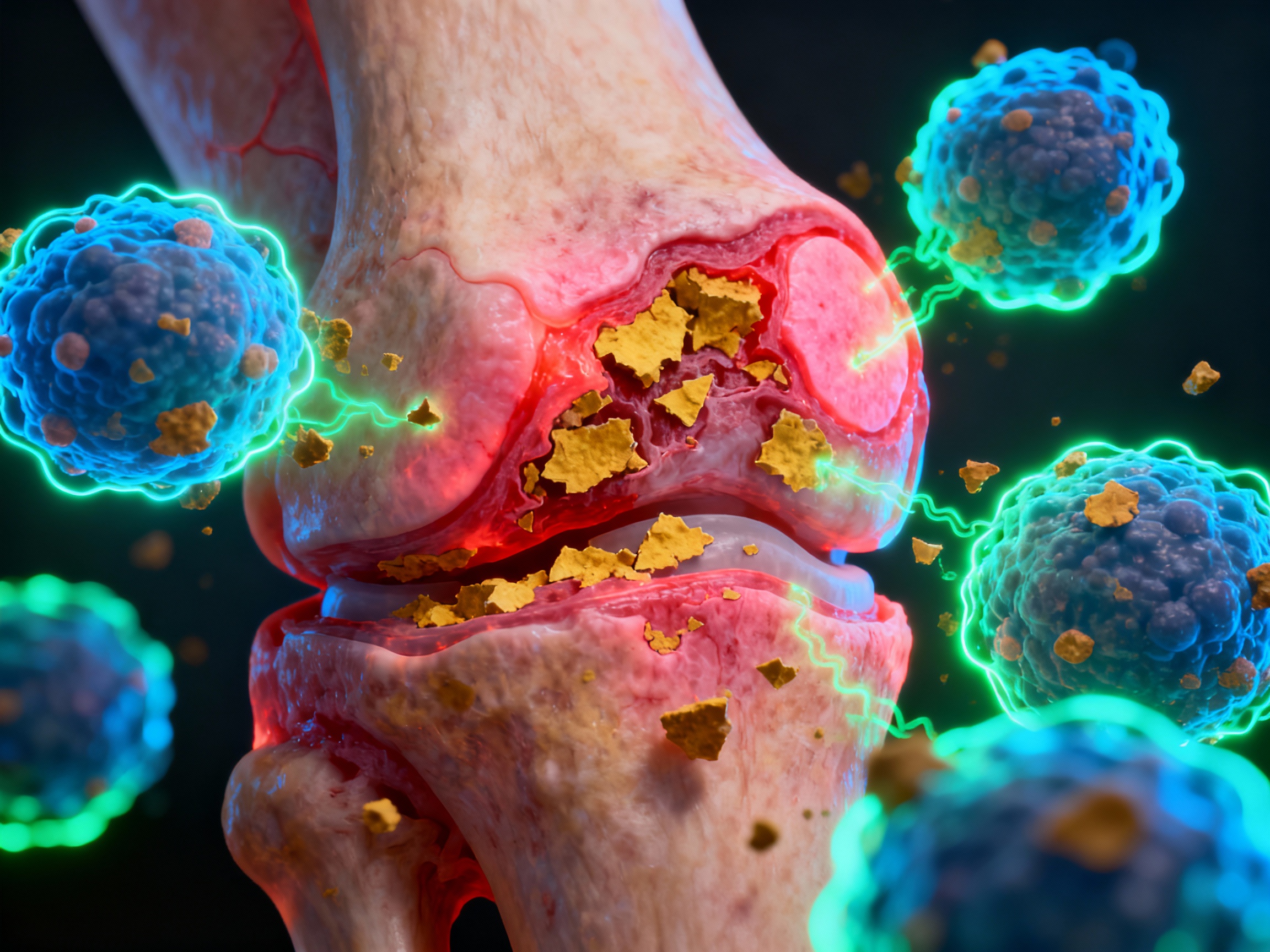
Synovial inflammation with immune cell infiltration destroying joint cartilage
Tph cells (CD4+PD1+CXCR5- T peripheral helper cells) accumulate in the synovium, generating IL-21 that drives B cell activation. These cells present citrullinated peptides - post-translationally modified self-proteins - to activate B cells that produce anti-citrullinated protein antibodies (ACPA). Macrophages infiltrate the synovium and release matrix metalloproteinases (MMPs) that destroy cartilage and bone. TNF-α, IL-6, and IL-1β create a pro-inflammatory environment, perpetuating the destructive cycle.
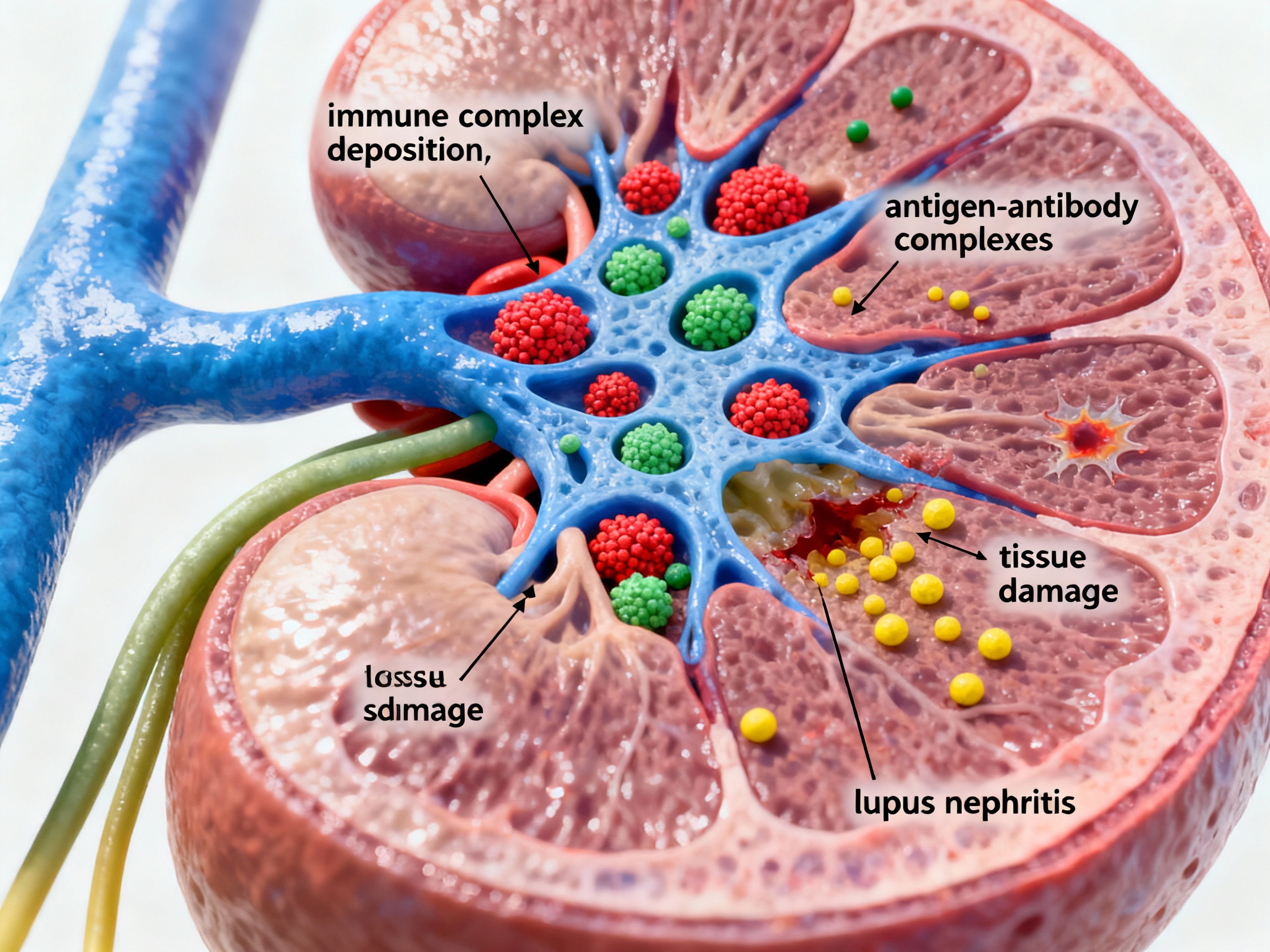
Immune complexes depositing in glomeruli causing lupus nephritis
In systemic lupus erythematosus, antigen-antibody immune complexes deposit in the glomeruli of the kidneys. These complexes activate the complement system, generating inflammatory mediators like C3a and C5a that recruit neutrophils and macrophages. The local inflammatory response damages the glomerular basement membrane, leading to proteinuria, hematuria, and progressive renal dysfunction. Type I interferons drive metabolic dysregulation that perpetuates the immune attack.
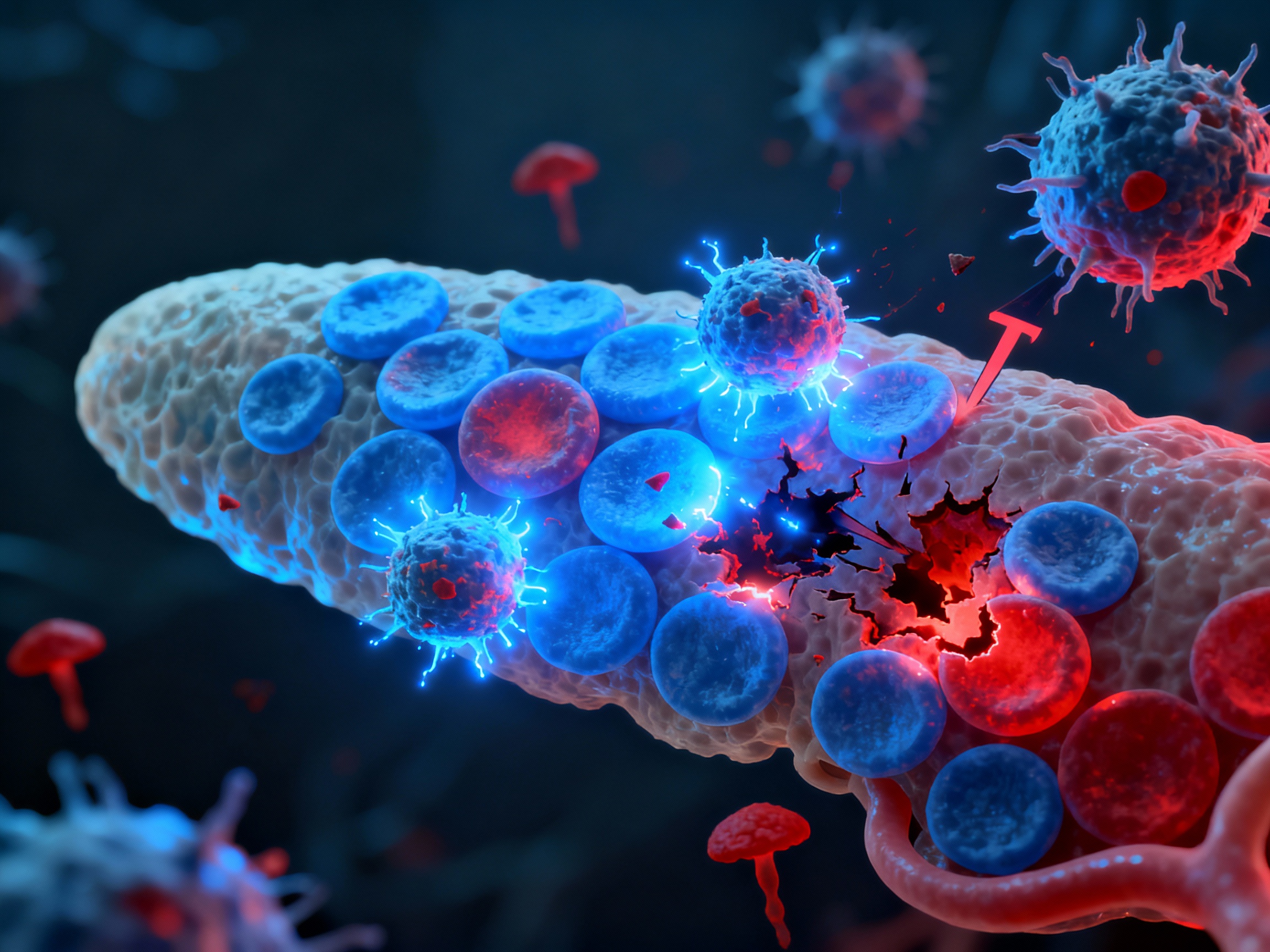
T cells infiltrating pancreatic islets and destroying insulin-producing beta cells
CD4+ and CD8+ T cells infiltrate pancreatic islets following chemokine gradients (CXCL10, CCL2). They differentiate into effector cells secreting IFN-γ and TNF-α. CD8+ cytotoxic T cells directly kill insulin-producing beta cells via perforin and granzyme. B cells assist by producing anti-β-cell antibodies (anti-GAD65, anti-IA-2, anti-insulin). Progressive beta cell destruction leads to absolute insulin deficiency.
Autoimmune disease disrupts cellular metabolism, creating a vicious cycle of energy failure and immune dysregulation.
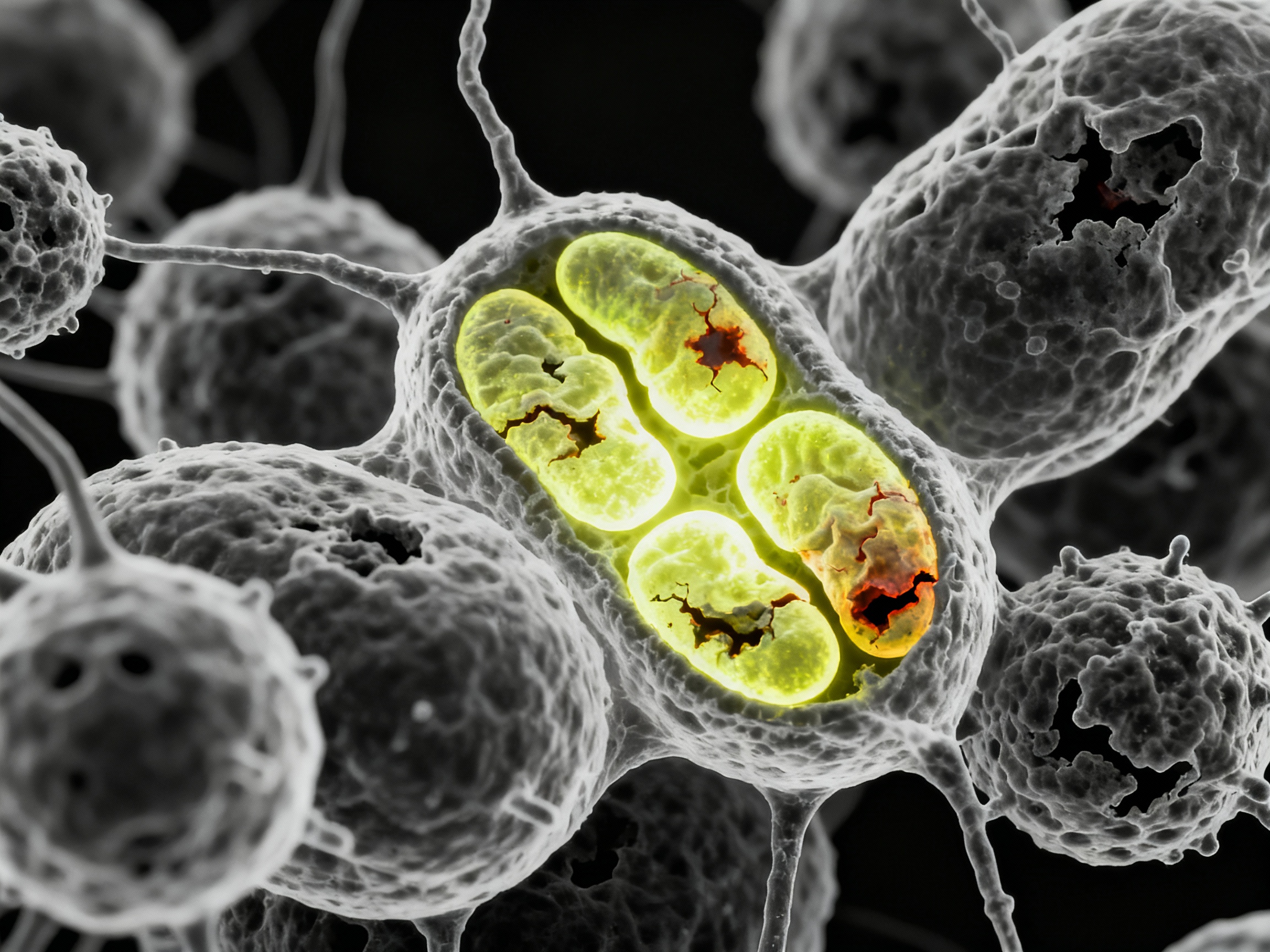
Mitochondrial dysfunction driving immune dysregulation and cellular energy crisis
Mitochondrial changes profoundly affect metabolism and immune response in systemic sclerosis, causing oxidative damage leading to cell and tissue injury in Sjögren's syndrome. Chronic inflammation impairs mitochondrial function, reducing ATP production and increasing reactive oxygen species (ROS) generation.
This metabolic reprogramming links cellular energy needs to immune dysfunction. Activated immune cells shift from oxidative phosphorylation to glycolysis (Warburg effect), demanding massive glucose uptake. This metabolic shift supports rapid proliferation and cytokine production but comes at the cost of cellular exhaustion.
Different metabolic patterns emerge at different disease stages in rheumatoid arthritis. In SLE, type I interferon-driven metabolic dysregulation affects T cell differentiation, proliferation, and cytokine secretion, perpetuating the autoimmune cascade.
When the immune system's brakes fail, nothing stops the autoimmune cascade.
Regulatory T cell dysfunction losing suppressive control over autoreactive immunity
Regulatory T cells (Tregs) marked by FOXP3 expression normally suppress autoreactive immune responses and maintain peripheral tolerance. They secrete anti-inflammatory cytokines (IL-10, TGF-β), consume IL-2 to starve effector T cells, and express CTLA-4 to outcompete CD28 for B7 binding on antigen-presenting cells.
In autoimmune disease, Treg function collapses. Reduced FOXP3 expression, impaired suppressive capacity, and resistance of effector T cells to suppression all contribute to loss of self-tolerance. The balance between regulatory cells and effector cells tips dramatically toward inflammation.
Without functional Tregs to restrain them, autoreactive T cells proliferate unchecked, B cells produce autoantibodies without restraint, and the cytokine storm intensifies. The immune system's fail-safe mechanisms have failed.
Autoimmune disease perpetuates itself through self-amplifying loops, each iteration causing more damage.
Click any stage in the cycle above to see detailed information
Autoimmune disease oscillates between periods of active inflammation and relative quiet.